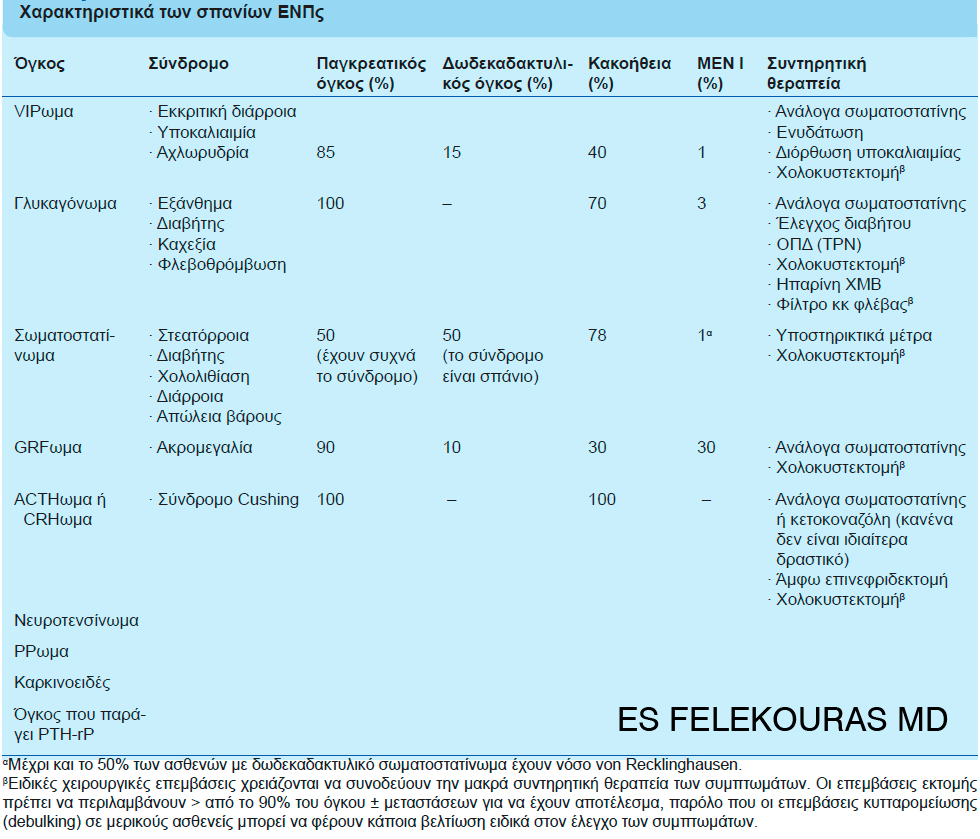
Όλα τα pNETs είναι σπάνια, αλλά αυτά εκτός του ινσουλινώματος και του γαστρινώματος είναι ακόμα πιο σπάνια (εικόνα 1). Η διάγνωση και η αντιμετώπισή τους είναι γενικά όπως όλων των pNETs, αλλά μερικά ειδικότερα στοιχεία όσον αφορά τη διάγνωση και τη θεραπεία αξίζει να αναφερθούν εδώ.
Εικόνα 1:
VIPόµα [σύνδροµο παγκρεατικής χολέρας ή σύνδροµο Verner-Morrison ή WDHA σύνδροµο (Watery Diarrhea, Hypokalemia, Achlorhydria)]
Το αγγειοδραστικό εντερικό πεπτίδιο (vasoactive intestinal peptide, VIP) είναι ένα νευροπεπτίδιο που ανευρίσκεται σε όλο το νευρικό σύστημα και ιδίως στο ΚΝΣ. Περιφερικά το VIP ανευρίσκεται στους πεπτιδικούς νευρώνες των αγγείων του σπλαγχνικού νευρικού συστήματος συμπεριλαμβανομένου αυτού του λεπτού και του παχέος εντέρου και του εξωκρινούς συστήματος του παγκρέατος. Το VIP ενεργεί μέσω του υποδοχέα της ειδικής αδενικής κυκλάσης (adenylate cyclase).
Οι νευροενδοκρινείς όγκοι που παράγουν VIP, είναι εξαιρετικά σπάνιοι, με συχνότητα ένα νέο περιστατικό ανά 10.000.000 πληθυσμού ετησίως. Εντοπίζονται σχεδόν αποκλειστικά (80-90%) στο πάγκρεας (συνήθως στην ουρά και στο σώμα), ενώ ένα 15% ανευρίσκεται στο δωδεκαδάκτυλο. Εξωπαγκρεατικά VIPώματα έχουν αναφερθεί στο οπίσθιο περιτόναιο, στο ήπαρ τον οισοφάγο και το λεπτό έντερο, όπως και σπάνιες περιπτώσεις που το σύνδρομο προέρχεται από καρκινοειδή του εντέρου, βρογχογενή καρκινώματα ή/και φαιοχρωμοκυτώματα που παράγουν VIP. Τα VIPώματα είναι συνήθως μονήρεις μεγάλοι όγκοι και σπάνια πολλαπλοί (4-9%) οπότε και συνδέονται με ΜΕΝ Ι. Στο 40-50% των περιπτώσεων είναι κακοήθεις και η μέση ηλικία διάγνωσης σε ενήλικες είναι τα 42-51 έτη, ενώ στα παιδιά τα 2-4 έτη.
Το σύνδρομο πρωτοαναγνωρίσθηκε το 1957 από τους Priest και Alexander και από τους Verner και Morrison το 1958 από όπου φέρει και το όνομα του αντίστοιχου συνδρόμου. Το σύνδρομο συνδέθηκε με παγκρεατικό όγκο που παράγει VIP το 1973. Όπως γνωρίζουμε και για τα άλλα pNETs υπάρχει μια διαφωνία αν το VIP είναι μια παγκρεατική ορμόνη που κατά μερικούς παράγεται από τα D2 κύτταρα και κατά άλλους, όπως και η γαστρίνη, απλώς ανευρίσκεται μόνο σε αυτούς τους όγκους των νησιδίων. Τα εκκριτικά κοκκία του VIP είναι μικρά (120-180 nm) και παρομοιάζουν με αυτά των D κυττάρων του φυσιολογικού εντέρου. Ούτε τα ιστολογικά ευρήματα ούτε αυτά του ηλεκτρονικού μικροσκοπίου μας επιτρέπουν να διαχωρίσουμε τα VIPώματα από άλλα pNETs, αλλά η ανοσοϊστοχημική ανεύρεση του VIP είναι ισχυρώς ενδεικτική του VIPόματος, μια και δεν ανευρίσκεται συχνά σε άλλα pNETs.
Η κλινική εικόνα του συνδρόμου είναι πολύ ευμετάβλητη. Σταθερά ευρήματα είναι η έντονη μεγάλου όγκου εκκριτική διάρροια (89-100%), η υποογκαιμία- αφυδάτωση (44-100%), η υποκαλιαιμία (67-100%) και η οξέωση με ασταθή ευρήματα την αχλωρυδρία ή υποχλωρυδρία, την υπερκαλιαιμία, την υπεργλυκαιμία και το ερυθηματώδες εξάνθημα του προσώπου και του κορμού (14-28%). Η διάρροια επιμένει και όταν ο ασθενής είναι νήστις (εκκριτική διάρροια) και παρόλη τη ρινογαστρική αναρρόφηση (που την διαφοροποιεί από το ΣΖΕ). Οι ημερήσιες απώλειες ύδατος και ηλεκτρολυτών είναι πολύ μεγάλες με ποσότητες μεγαλύτερες από 1 L και τις περισσότερες φορές περισσότερο από 3 L/ημερησίως, γι’ αυτό δε ονομάζεται και σύνδρομο παγκρεατικής χολέρας. Συχνά οι ασθενείς παρουσιάζουν απώλεια βάρους και κοιλιακό άλγος.
Η παθογένεια της υποκαλιαιμίας είναι αυτή της απώλειας από τη διάρροια, αλλά και λόγω του δευτεροπαθούς υπεραλδοστερινισμού λόγω διέγερσης της απελευθερούμενης ρενίνης από το VIP. Άλλες ηλεκτρολυτικές διαταραχές που συνοδεύουν το σύνδρομο είναι η υπερασβεστιαιμία με πιθανή αιτία τη διέγερση από το VIP της οστεολυτικής δραστηριότητας. Η υπεργλυκαιμία αποδίδεται στη γλυκογονολυτική δράση του VIP στο ήπαρ. Το εξάνθημα είναι αποτέλεσμα των αγγειοδιασταλτικών δραστηριοτήτων του VIP, όπως και η ύπο- αχλωρυδρία είναι αποτέλεσμα των ανασταλτικών επί της γαστρικής έκκρισης ιδιοτήτων του VIP.
Οι βιοχημικές εξετάσεις επιβεβαιώνουν την κλινική εικόνα. Η ανευρισκόμενη υποκαλιαιμία είναι συχνά βαριά (Κ+ <2,5 mmol/L), ενώ η υπερασβεστιαιμία, και η υπεργλυκαιμία δεν είναι ιδιαίτερα σοβαρές.
Η διαγνωστική τριάδα στο σύνδρομο Verner- Morrison είναι η μεγάλης ποσότητας εκκριτική διάρροια, τα υψηλά επίπεδα κυκλοφορούντος VIP και ενός παγκρεατικού όγκου ή άλλου όγκου συνδεμένου με την παραγωγή του VIP. Ποσότητα κοπράνων μικρότερη των 700 ml/ημερησίως θεωρείται ότι αποκλείει τη διάγνωση του VIPόματος. Η διάγνωση τίθεται με την ανίχνευση υψηλών τιμών VIP στον ορό (συνήθως πάνω από 200 pg/ml).
Οι συνήθεις παθήσεις από όπου το σύνδρομο πρέπει να διαφοροδιαγιγνώσκεται μια και δημιουργούν μια εικόνα ψεύδο- VIPόματος, είναι οι κάτωθι:
- κατάχρηση υπακτικών (ιστορικό)
- βακτηριακή και παρασιτική διάρροια (καλλιέργειες)
- καρκινοειδές σύνδρομο (αυξημένα επίπεδα 5-υδροξυινδολο-οξικού οξέος στα ούρα)
- ΣΖΕ (αυξημένη γαστρίνη), και
- εκκριτική διάρροια άγνωστης αιτιολογίας
Η ανατομική εντόπιση του όγκου συνήθως γίνεται με τη βοήθεια της ΑΤ ή/και της MRI, μια και οι όγκοι αυτοί κατά τη διάγνωση είναι αρκετών cm σε διάμετρο. Φυσικά η αγγειογραφία και το SRS έχουν κάποιο ρόλο στη διάγνωση ως συμπληρωματικές εξετάσεις. Το τελευταίο ειδικά βοηθά τα μέγιστα στην ανίχνευση λανθάνουσας μεταστατικής νόσου. Το VIPομα παρουσιάζει επιθετική συμπεριφορά, με ανεύρεση μεταστάσεων στο ήπαρ και τους λεμφαδένες σε ποσοστό 37-78% σε διάφορες μελέτες κατά τη στιγμή της διάγνωσης.
Σε παιδιά μικρότερα των 10 ετών και σπάνια στους ενήλικες (5% των περιπτώσεων), το σύνδρομο του VIPόματος προέρχεται από γαγγλιονεύρωμα ή γαγγλιονευροβλάστωμα εξωπαγκρεατικής προέλευσης και είναι λιγότερο κακοήθη (10%) από τα παγκρεατικά VIPώματα.
Θεραπεία
Άμεσα η θεραπεία αρχίζει με τη διόρθωση της υποογκαιμίας και της αφυδάτωσης καθώς και των ηλεκτρολυτικών διαταραχών και της οξέωσης (υπερχλωραιμική χωρίς χάσμα ανιόντων). Μόλις η θεραπεία αυτή αρχίσει, άμεσα θα πρέπει να αρχίσει και η θεραπεία με ανάλογα της σωματοστατίνης (οκτρεοτίδη ή λαντρεοτίδη) ή με αυτά μακράς δράσεως. Με τη θεραπεία αυτή η συγκέντρωση του VIP στο πλάσμα βελτιώνεται στο 80-89% των ασθενών, όπως και η διάρροια άμεσα και σε απώτερο χρόνο στο 78-87% των ασθενών με VIPόμα. Σε ασθενείς που δεν αντιδρούν στη θεραπεία αυτή ή αν τα συμπτώματα υποτροπιάσουν παρά την αύξηση της δόσης, συνιστάται η σύγχρονη χορήγηση γλυκοκορτικοειδών.
Χειρουργική θεραπεία
Όπως και για τους λοιπούς νευροενδοκρινείς όγκους του παγκρέατος η χειρουργική θεραπεία είναι η αντιμετώπιση εκλογής. Στόχος της είναι η εξαίρεση όλης της βλάβης. Έτσι μετά από τις διαγνωστικές εξετάσεις, όλοι οι υποψήφιοι για εκτομή (με και χωρίς μεταστατική νόσο) που είναι δυνατόν να υποβληθούν σε χειρουργική επέμβαση, θα πρέπει να υποβληθούν σε ερευνητική λαπαροτομία για πιθανή εκτομή R0, και με συνοδό λεμφαδενικό καθαρισμό. Όλοι οι ασθενείς πρέπει να υποβληθούν σε χολοκυστεκτομή ανεξαρτήτως σταδίου νόσου, μια και η επέμβαση αυτή διευκολύνει τη θεραπεία με ανάλογα σωματοστατίνης.
Τα περισσότερα VIPώματα μπορεί να εξαιρεθούν με περιφερική παγκρεατεκτομή. Στους ασθενείς αυτούς και ειδικά σε αυτούς με μεταστατική νόσο, οι επεμβάσεις εκτομής πρέπει να περιλαμβάνουν την αφαίρεση περισσότερο από το 90% του όγκου για να έχουν αποτέλεσμα, παρόλο που οι επεμβάσεις κυτταρομείωσης (debulking) σε μερικούς ασθενείς μπορεί να έχουν κάποια βελτίωση ειδικά στην κλινική εικόνα του συνδρόμου. Τα επινεφρίδια και ο οπισθοπεριτοναϊκός χώρος πρέπει να διερευνηθούν προσεκτικά, εάν κανένας παγκρεατικός όγκος δεν ανευρίσκεται.
Θεραπεία μπορεί να επιτευχθεί στο 30% των ασθενών και η κλινική εικόνα να υφεθεί πλήρως σε ένα άλλο 33%. Η επιβίωση και ο έλεγχος των συμπτωμάτων (78%) είναι καλύτερα για τα γαγγλιονευροβλαστώματα που παράγουν VIP.
Στους ασθενείς με ανεγχείρητα VIPώματα η θεραπεία είναι όπως αναφέρθηκε τα ανάλογα της σωματοστατίνης, που είναι χρήσιμα στον έλεγχο της διάρροιας, και η χημειοθεραπεία, που δυστυχώς όμως είναι σπάνια αποτελεσματική.
Γλουκαγόνωµα ή Γλυκαγόνωµα
Το γλουκαγόνωμα είναι ένας αρκετά σπάνιος όγκος με συχνότητα ένα νέο περιστατικό ανά 5.000.000 πληθυσμού ανά έτος και παρουσιάζεται κατά τη μέση ή μεγάλη ηλικία με ελαφρά προτίμηση στις γυναίκες. Ενδεχομένως, όμως, τα γλυκαγονώματα μπορεί να είναι πιο συχνά, μιας και σε νεκροτομικές σειρές σε διαβητικούς τύπου ΙΙ, η συχνότητα ανεύρεσης φθάνει στο 0,8% (μικρογλουκαγονώματα). Οφείλεται στη νεοπλασία των α κυττάρων των νησιδίων του παγκρέατος που παράγουν γλυκαγόνο και άλλα πεπτίδια. Το σύνδρομο περιγράφηκε αρχικά το 1942 από δερματολόγους (Becker και συν.) που διέγνωσαν τη σχέση μεταξύ ενός παγκρεατικού όγκου και μιας δύσκολης στη θεραπεία δερματίτιδας. Οι περισσότεροι όγκοι είναι σποραδικοί. Περίπου το 5-17% συνδέονται με το σύνδρομο ΜΕΝ I. Σε ασθενείς που έχουν σποραδικό γλυκαγόνωμα η ηλικία παρουσίασής του είναι η πέμπτη δεκαετία της ζωής, ενώ όταν συνδέεται με ΜΕΝ Ι είναι μικρότερη. Λόγω των μη ειδικών συμπτωμάτων που προκαλεί ο όγκος, συχνά η διάγνωση καθυστερεί αρκετά, με αποτέλεσμα ο όγκος να έχει συνήθως αρκετά μεγάλο μέγεθος κατά τη διάγνωσή του (μέσος όρος 5 cm). Οι όγκοι αυτοί εμφανίζονται σχεδόν αποκλειστικά στο πάγκρεας ως μονήρη νεοπλάσματα με μικρή προτίμηση στην ουρά του οργάνου, αν και έχει αναφερθεί επίσης σειρά ασθενών όπου το 10-12% είχε πολλαπλούς όγκους ή διάχυτη κατάληψη του οργάνου από μια ενιαία μάζα. Τα σποραδικά γλυκαγονώματα είναι κακοήθη σε ποσοστό >80% με συνήθεις μεταστάσεις στο ήπαρ και τους λεμφαδένες κατά τη στιγμή της διάγνωσης.
Η κλινική συμπτωματολογία του συνδρόμου του γλουκαγονώματος είναι αποτέλεσμα της υπερπαραγωγής γλουκαγόνου αν και όχι σε όλο το φάσμα των κλινικών εκδηλώσεών του. Οι ασθενείς συνήθως πάσχουν από αναιμία, απώλεια βάρους (70-80%), βαριά καχεξία και υπολευκωματιναιμία λόγω του έντονου καταβολισμού των πρωτεϊνών, ο οποίος οφείλεται στην ανεξέλεγκτη νεογλυκογένεση. Συνήθη συμπτώματα είναι επίσης ο ήπιος σακχαρώδης διαβήτης (75%), η δερματίτιδα (65-80%), η εν τω βάθει φλεβοθρόμβωση των κάτω άκρων με συχνή εμβολική νόσο και η διαταραχή της προσωπικότητας (στην αγγλική βιβλιογραφία αναφέρονται τα τέσσερα D, από τα αρχικά των λέξεων diabetes, dermatitis, deep-vein thrombosis, depression). Η διάρροια (15-30%) και το κοιλιακό άλγος συνυπάρχουν λιγότερο συχνά. Η δερματίτιδα του συνδρόμου χαρακτηρίζεται από το λεγόμενο νεκρωλυτικό μεταναστευτικό ερύθημα. Αυτό συνίσταται σε ένα μικρό εξάνθημα που συνήθως ξεκινά από τους βουβώνες και εξελίσσεται μέσα σε 7-14 ημέρες επεκτεινόμενο προς το περίνεο και στα κάτω άκρα. Στη συνέχεια μεταναστεύει και γίνεται κνιδωτικό ερυθρό εξάνθημα με έντονο κνησμό κυρίως γύρω από τους οφθαλμούς (Εικόνα 1), συνοδεύεται δε από στοματίτιδα, γλωσσίτιδα και χειλίτιδα (30-40%). Το γλυκαγόνωμα βρέθηκε ότι συνδέεται με ένα χαμηλό επίπεδο αμινοξέων, των οποίων η παρεντερική χορήγηση αποδείχθηκε ότι επιφέρει σχεδόν πλήρη εξαφάνιση του εξανθήματος, χωρίς να αλλάζουν τα επίπεδα του γλουκαγόνου στον ορό. Ένα σύνδρομο ψευδογλουκαγονώματος έχει περιγραφεί σε ασθενείς που έχουν νεκρωλυτικό μεταναστευτικό ερύθημα χωρίς παγκρεατικό όγκο (από άγνωστη αιτία), σε ασθενείς με σοβαρή χρόνια νόσο (ηπατική νόσο, παγκρεατίτιδα, κακοήθεια κ.ά.). Μόνο ελάχιστοι εκ των ασθενών αυτών παρουσιάζουν υψηλά επίπεδα γλουκαγόνου.

Εικόνα 1: Νεκρωλυτικό μεταναστευτικό ερύθημα προσώπου σε ασθενή με γλυκαγόνωμα.
Η διάγνωση του συνδρόμου στηρίζεται στο χαρακτηριστικό εξάνθημα, την ανίχνευση υψηλών τιμών γλουκαγόνου του ορού (με ή χωρίς πρόκληση με σεκρετίνη), οι οποίες συνήθως είναι πάνω από 1000 pg/ ml και συνήθως 200-2000 pg/ml (ΦΤ: 150-190 pg/ml) και ένα παγκρεατικό όγκο. Η ανατομική εντόπιση του όγκου είναι ευχερής λόγω του μεγάλου μεγέθους του και η ΑΤ τις περισσότερες φορές είναι η μόνη εξέταση που απαιτείται. Η MT μπορεί να μας δώσει τα ίδια και ίσως καλύτερα αποτελέσματα. Σε μερικές σειρές μέχρι και το 20% των ασθενών με γλυκαγόνωμα έχουν συγχρόνως και ΣΖΕ, ενώ το 13-17% έχουν ΜΕΝ I.
Μόλις τεθεί η διάγνωση, ο ασθενής πρέπει να προετοιμαστεί με τη χορήγηση ολικής παρεντερικής διατροφής που περιέχει αμινοξέα, με ταυτόχρονη χορήγηση αναλόγων σωματοστατίνης (SSA) για συμπτωματική ανακούφιση. Η προφύλαξη με ηπαρίνη χαμηλού μοριακού βάρους πριν από την επέμβαση είναι απαραίτητη, μια και το ένα τρίτο αυτών των ασθενών έχει αναφερθεί ότι περιπλέκεται με περιεγχειρητική φλεβοθρόμβωση και ένα 11% με εμβολική νόσο.
Η χειρουργική εξαίρεση του όγκου αποτελεί τη μοναδική πιθανότητα για ίαση. Συνήθως, λόγω της εντόπισης και του μεγέθους του όγκου, απαιτείται περιφερική παγκρεατεκτομή. Σχεδόν όλα τα γλυκαγονώματα είναι κακοήθη γι’ αυτό και απαιτείται επιθετική χειρουργική τακτική (Εικόνα2). Εάν υπάρχουν λεμφαδενικές ή ηπατικές μεταστάσεις αυτές θα πρέπει να συναφαιρούνται με τον κύριο όγκο. Δυστυχώς κάτι τέτοιο είναι εφικτό μόνο στο 30% των ασθενών. Στις υπόλοιπες περιπτώσεις αρκετοί ερευνητές υποστηρίζουν την αφαίρεση του μεγαλύτερου μέρους του όγκου (debulking). Με αυτόν τον τρόπο διευκολύνεται η συμπτωματική αντιμετώπιση των λειτουργικών διαταραχών του συνδρόμου με την προσθήκη SSA. Η χημειοθεραπεία είναι απογοητευτική για τη μεταστατική νόσο. Παρόλα αυτά, όμως, οι ασθενείς με γλυκαγόνωμα έχουν αρκετά καλή επιβίωση.
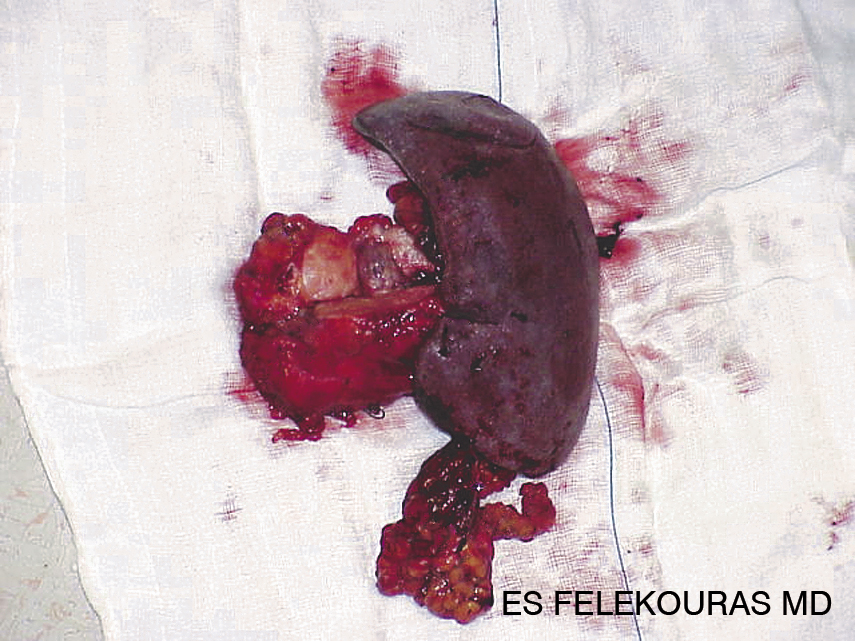
Εικόνα 2: Περιφερική παγκρεατεκτομή για γλουκαγόνωμα.
Σωµατοστατίνωµα
Το σωματοστατίνωμα αποτελεί σπάνιο ορμονοεκκριτικό όγκο του παγκρέατος. Περιγράφηκε πρώτη φορά το 1977, ενώ το πλήρες σύνδρομο (στεατόρροια, διαβήτης, υποχλωρυδρία και χολολιθίαση) αναγνωρίσθηκε 2 έτη αργότερα. Σε μια μελέτη που έγινε το 1999 από Ιάπωνες ερευνητές, περιεγράφηκαν συνολικά μέχρι τότε 173 περιστατικά παγκοσμίως. Στα 81 από αυτά η βλάβη εντοπιζόταν στο πάγκρεας και στα 92 στο δωδεκαδάκτυλο. Υπολογίζεται ότι η ετήσια επίπτωση της νόσου είναι ένα νέο περιστατικό ανά 40.000.000 πληθυσμού. Το σωματοστατίνωμα είναι ένα PNET που εκφράζει σωματοστατίνη με ανοσοϊστοχημεία ενώ μπορεί να εμφανιστεί με (11%) ή χωρίς (89%) το σύνδρομο του σωματοστατινώματος. Ο ορισμός αυτός πάντως δεν χρησιμοποιείται από όλους. Στην πραγματικότητα, σε πολλές μελέτες ο όρος σωματοστατίνωμα χρησιμοποιείται για όλους τους νευροενδοκρινείς όγκους που εκφράζουν με ανοσοϊστοχημεία σωματοστατίνη, ενώ δεν απαιτείται η ύπαρξη συνοδευτικού λειτουργικού συνδρόμου.4
Τα παγκρεατικά σωματοστατινώματα συνήθως είναι μεγάλα (πάνω από 5 cm), τείνουν δε να εντοπίζονται στην κεφαλή του οργάνου (κεφαλή:σώμα:ουρά= 30:3:11). Οι όγκοι εκτός παγκρέατος εντοπίζονται κυρίως στο δωδεκαδάκτυλο (90%) αλλά και αλλαχού όπως στη θηλή του Vater, τη νήστιδα και τον κυστικό πόρο, ίσως λόγω του μεγάλου αριθμού των D κυττάρων που παράγουν σωματοστατίνη στις περιοχές αυτές. Πολλά από τα δωδεκαδακτυλικά νεοπλάσματα συχνά σχετίζονται με τη νόσο von Recklinghausen (NF-1). Το 50% των ασθενών με σωματοστατίνωμα έχει και άλλες ενδοκρινοπάθειες, όπως π.χ. MEN Ι και MEN II.
Οι παγκρεατικοί όγκοι είναι μονήρεις (90-96%) δια- μέτρου 1,5-10 cm. Είναι κακοήθεις σε ποσοστό 70-90%, οι δε μεταστάσεις είναι συχνότερες στο ήπαρ, στους λεμφαδένες και στα οστά. Ίσως τα παγκρεατικά σωματοστατινώματα να μεθίστανται πιο εύκολα στο ήπαρ από τα υπόλοιπα. Οι δωδεκαδακτυλικοί όγκοι είναι συνήθως πολύ μικρότεροι κατά τη διάγνωση, μπορούν δε να παρουσιασθούν λόγω θέσεως με απόφραξη του ανωτέρου πεπτικού (ΑΑΠ) ή αποφρακτικό ίκτερο.
Η κλινική παρουσίαση του σωματοστατινώματος είναι απρόβλεπτη. Μερικοί ασθενείς παρουσιάζονται με αποφρακτικό ίκτερο που προκαλείται από την πίεση του όγκου στα εξωηπατικά χοληφόρα, κοιλιακό άλγος, απώλεια βάρους κ.λπ. (μη ειδικά συμπτώματα της μάζας του όγκου), ενώ άλλοι έχουν διάρροια και χολολιθίαση (συμπτώματα του συνδρόμου). Οι όγκοι εμφανίζονται στην ηλικία των 40-60 ετών. Στις περισσότερες περιπτώσεις τα σωματοστατινώματα βρίσκονται κατά τη διάρκεια της λαπαροτομίας για χολοκυστεκτομή ή κατά τη διάρκεια μελετών απεικόνισης του γαστρεντερικού για διάφορα μη ειδικά συμπτώματα, όπως ο κοιλιακός πόνος ή η διάρροια.
Το σύνδρομο που προκαλεί η υπερέκκριση σωματοστατίνης από τους όγκους αυτούς, ονομάζεται και ανασταλτικό (inhibitory syndrome). Εμφανίζεται κλινικά σε ποσοστό που δεν ξεπερνά το 20% των παγκρεατικών σωματοστατινωμάτων συγκριτικά με το 2,5% των δωδεκαδακτυλικών. Η παθοφυσιολογία του συνδρόμου μπορεί να εξηγηθεί από τις γνωστές δράσεις της σωματοστατίνης. Χαρακτηρίζεται δε από την εμφάνιση κοιλιακού άλγους, χολολιθίασης (68%), απώλειας βάρους, διάρροιας (60%), στεατόρροιας (47%), διαβήτη (95%) και υποχλωρυδρίας (26%). Σε ένα 10% παρουσιάζονται συμπτώματα υπογλυκαιμίας που αποδεικνύει τις απρόβλεπτες αλληλοεπιδράσεις των ορμονών. Σε μια μελέτη το 20% των ασθενών διεγνώσθησαν λανθασμένα ως έχοντες ινσουλίνωμα λόγω υπογλυκαιμίας.
Η διάγνωση του συνδρόμου τίθεται με την ανίχνευση στον ορό υψηλών τιμών σωματοστατίνης. Οι μέτριες αυξήσεις των τιμών της σωματοστατίνης πρέπει να ερμηνεύονται με προσοχή, επειδή μπορούν να εμφανιστούν και σε άλλες ΝΕ διαταραχές όπως το μυελοειδές καρκίνωμα του θυρεοειδούς, το μικροκυτταρικό καρκίνωμα πνεύμονος, το φαιοχρωμοκύτωμα και άλλα εξω επινεφριδιακά παραγαγγλιώματα που παράγουν κατεχολαμίνες.
Έτσι εάν η διάγνωση δεν τεθεί προεγχειρητικά τότε τίθεται με την ιστολογική εικόνα του pNET όπου ανευρίσκεται σωματοστατίνη με ανοσοϊστοχημικές μεθόδους. Η ανεύρεση σε αυτές τις περιπτώσεις υψηλών τιμών σωματοστατίνης στον ορό είναι απαραίτητη για να τεθεί η διάγνωση του λειτουργικού συνδρόμου. Τα επίπεδα σωματοστατίνης πλάσματος είναι συνήθως υψηλά στο παγκρεατικό σωματοστατίνωμα, ενώ στους δωδεκαδακτυλικούς ή μικρούς εντερικούς όγκους τα επίπεδα του πλάσματος μπορούν να είναι κανονικά.
Αυτή τη στιγμή το κλειδί για τη διάγνωση του συνδρόμου του σωματοστατινώματος είναι:
- η αναγνώριση της κλινικής εικόνας
- ο έλεγχος επιπέδων σωματοστατίνης σε ασθενείς με τα ακόλουθα ευρήματα:
a. διαβήτης χωρίς οικογενειακό ιστορικό
b.χολολιθίαση
c. παγκρεατική μάζα
d. ανεξήγητη διάρροια
Όπως έχει ήδη αναφερθεί τα δωδεκαδακτυλικά σωματοστατινώματα συνδέονται σε μεγάλο ποσοστό με τη νόσο του von Recklinghausen (NF1). Αυτά (επί NF1) προσομοιάζουν με τα σποραδικά δωδεκαδακτυλικά σωματοστατινώματα, δηλαδή σπάνια εμφανίζουν το σύνδρομο (2%), η δε σωματοστατίνη στον ορό δεν είναι συνήθως αυξημένη, και όπως και τα σποραδικά παρουσιάζουν στην ιστολογική τους εικόνα ψαμμώματα (psammoma bodies). Ευτυχώς όμως είναι πιο καλοήθη από τα σποραδικά (53% vs 31%).
Ο εντοπισμός του όγκου δεν αποτελεί πρόβλημα, γίνεται δε με την ΑΤ ή/και MRI και σπάνια αγγειογραφία, μια και οι όγκοι είναι συνήθως μεγάλοι. Το SRS δεν προσθέτει στη διάγνωση μια και τα σωματοστατινώματα δεν διαθέτουν υποδοχείς σωματοστατίνης αν και οι απόψεις επί αυτού διίστανται.
Θεραπεία
Δεν υπάρχει συντηρητική θεραπεία άλλη από την υποστηρικτική και από τη χορήγηση αναλόγων σωματοστατίνης. Μόνο η χειρουργική έχει να προσφέρει σε αυτούς τους πάσχοντες. Ασθενείς με εντοπισμένη νόσο πρέπει να υποβληθούν σε ανατομική εκτομή, συνήθως επέμβαση Whipple, λόγω της εντόπισης των όγκων αυτών στην κεφαλή του παγκρέατος. Η πενταετής επιβίωση στους ασθενείς χωρίς μεταστάσεις μετά από χειρουργική θεραπεία είναι 100%, που είναι σημαντικά καλύτερη από την επιβίωση 60% σε ασθενείς με μεταστάσεις. Οι ηπατικές μεταστάσεις είναι συχνές. Παρ’ όλο που οι επεμβάσεις κυτταρομείωσης είναι για πολλούς απαραίτητες, σαφώς δεν έχει αποδειχθεί ότι το debulking παρατείνει την επιβίωση. Τα μικρά δωδεκαδακτυλικά σωματοστατινώματα μπορεί να θεραπευθούν με τοπική εκτομή. Πρέπει να σημειωθεί ότι ενδείκνυται ταυτόχρονη χολοκυστεκτομή, ανεξαρτήτως της ύπαρξης ή όχι χολολιθίασης, μιας και τελικά αυτή θα επισυμβεί λόγω της χορήγησης σωματοστατίνης.
Ακόµα πιο σπάνια pNETs (pNETs µε παραγωγή έκτοπης ορµόνης)
Διάφορα λειτουργικά pNETs που συνοδεύονται από το αντίστοιχο ορμονοπαραγωγό σύνδρομο έχουν αναφερθεί στη διεθνή βιβλιογραφία, όπως:
- αυτά που εκκρίνουν GRF (GRFόματα)
- αυτά που εκκρίνουν νευροτενσίνη (Neurotensinomas)
- αυτά που εκκρίνουν ACTH (ACTHόμα) ή CRH (GRFόματα)
- αυτά που εκκρίνουν PTH-RP (parathyroid hormone- related peptide)
- αυτά που εκκρίνουν Pancreatic Polypeptide (PP)
GRFόµα
Τα GRFόματα είναι σπάνιοι όγκοι (έχουν ανακοινωθεί 50 περιπτώσεις) που εκκρίνουν υπερβολικές ποσότητες growth hormone releasing factor (GRF). Πρώτη φορά περιεγράφηκαν το 1982, η δε κλινική τους εικόνα είναι αυτή της ακρομεγαλίας.
Τα GRFόματα συνδέονται στενά με ΜΕΝ I: 30% ανευρίσκονται στο πάγκρεας και ειδικά στην ουρά, 50% στον πνεύμονα, 10% στο λεπτό έντερο και τα επινεφρίδια, ενώ σε ένα 30% είναι πολλαπλά. Μεγάλο ποσοστό (40%) των όγκων αυτών συνυπάρχει με ΣΖΕ (συνύπαρξη με γαστρινώματα), ενώ 40% συνυπάρχουν με σύνδρομο Cushing.
Τα GRFόματα είναι συνήθως μεγάλα (>6 cm) και έχουν μεταστάσεις στο 33-39%, συνήθως στους λεμφαδένες και στο ήπαρ. Όπως και στα άλλα pNETs η ανίχνευση του GRF γίνεται με ανοσοϊστοχημεία στο ιστολογικό παρασκεύασμα.
Το κλινικό σύνδρομο που προκαλείται από τον όγκο αυτό προέρχεται από τις ιδιότητες του GRF. Το GRF είναι ένα πεπτίδιο 44 αμινοξέων που είναι ένας διεγέρτης της αυξητικής ορμόνης. Ως συνέπεια αυτού, οι ασθενείς έχουν το κλινικό σύνδρομο της ακρομεγαλίας. Οι όγκοι αυτοί παρουσιάζονται σε ασθενείς μέσης ηλικίας 40 ετών. Οι ασθενείς με εντερικά GRFόματα είναι νεότεροι. Οι γυναίκες (73%) παρουσιάζουν πιο συχνά τους όγκους αυτούς, ειδικά όταν πρόκειται περί παγκρεατικού όγκου (78%).
Η κλινική εικόνα του συνδρόμου είναι αποτέλεσμα:
- της ακρομεγαλίας (GRF, GH)^ οι ασθενείς εμφανίζουν μεγάλα άκρα, αλλαγές προσώπου και δέρματος, κεφαλαλγία και παγίδευση περιφερικών νεύρων
- των κλινικών συνδρόμων από υπερπαραγωγή άλλων ορμονών εκτός των ως άνω, δηλαδή μπορεί να συνυπάρχουν ΣΖΕ (γαστρίνωμα), σύνδρομο Cushing, υπογλυκαιμία (υπερινσουλιναιμία), υπερπρολακτιναιμία κ.ά., και τέλος
- των τοπικών κλινικών σημείων από την ανάπτυξη της μάζας.
Έτσι η διάγνωση του GRFόματος ως PNET είναι πιθανή σε κάθε ασθενή με:
- ακρομεγαλία χωρίς αδένωμα υπόφυσης
- ακρομεγαλία με συνοδό υπερπρολακτιναιμία
- μια παράδοξη αύξηση της αυξητικής ορμόνης σε χορήγηση TRH ή σε δοκιμασία γλυκόζης, και
- με ακρομεγαλία και ενδοκοιλιακή, παγκρεατική ή εντερική μάζα
Η ανεύρεση GRFόματος σε ασθενή με ακρομεγαλία παραμένει μια σπάνια νοσολογική οντότητα (1/177 ασθενείς). Η διάγνωση τίθεται με την ανεύρεση υψηλών επιπέδων αυξητικής ορμόνης στον ορό (συνήθως >5 μg/L άνδρες και >10 μg/L στις γυναίκες) και με σύγχρονη ανίχνευση αυξημένων επιπέδων GRF (GRF-IR επίπεδα >300 pg/ml), σε συνδυασμό βέβαια με την ανεύρεση μάζας στο πάγκρεας ή εξωπαγκρεατικά όπως αναφέρθηκε προηγουμένως.
Η ανίχνευση του όγκου και των πιθανών μεταστάσεων γίνεται όπως και στα άλλα pNETs. Η θεραπεία είναι χειρουργική με εκτομή του όγκου και των μεταστάσεων σε μια προσπάθεια να επιτευχθεί R0 εκτομή. Πριν από τη χειρουργική επέμβαση καλό είναι να γίνεται προσπάθεια μείωσης των επιπέδων των ορμονών με την χορήγηση αναλόγων της σωματοστατίνης, παρ’ όλο που είναι δύσκολο να επιτευχθεί απόλυτη ομαλοποίηση αυτών. Από μόνη της η χειρουργική επέμβαση επιτυγχάνει την υποχώρηση του συνδρόμου σε μικρό ποσοστό, αλλά η ακριβής πρόγνωση των όγκων αυτών δεν είναι γνωστή.
ACTHόµα ή CRHόµα
PNETs που παράγουν ACTH ή CRH ανευρίσκονται σπάνια και σχεδόν αποκλειστικά στο πάγκρεας (έχουν ανακοινωθεί 110 περιπτώσεις) και όχι στο δωδεκαδάκτυλο. Ασθενείς με τέτοιους όγκους συνήθως εμφανίζουν και άλλους ΝΕ όγκους και κυρίως ΣΖΕ. Φυσικά υπάρχουν και άλλα ΝΕ νεοπλάσματα που παράγουν ACTH όπως αυτά των βρόγχων.
Οι ασθενείς αυτοί παρουσιάζουν κλινικό σύνδρομο Cushing (το οποίο συμβαίνει στο 5% όλων των ασθενών με ΣΖΕ και στο 20% των ασθενών με ΣΖΕ και ΜΕΝ I).
Το 4-16% των περιπτώσεων του έκτοπου συνδρόμου Cushing οφείλεται σε ένα pNET. Σύνδρομο Cushing ανευρίσκεται στο 19% των ασθενών με γαστρίνωμα και ΜΕΝ I και στο 4-5% των περιπτώσεων σποραδικού γαστρινώματος, αλλά στους ασθενείς αυτούς η εστία δεν είναι στο πάγκρεας αλλά πρόκειται για ένα αδένωμα της υπόφυσης. Σε μια μελέτη η ανάπτυξη έκτοπου συνδρόμου Cushing σε ασθενή με PNETs βρέθηκε να παρουσιάζεται μόνο σε ύπαρξη μεταστατικής νόσου.
Το σύνδρομο Cushing σε ασθενείς με ACTHόμα ή CRHόμα είναι πολύ έντονο. Οι όγκοι αυτοί αυξάνονται πολύ γρήγορα σε μέγεθος, εμφανίζουν σχεδόν στην πλειοψηφία τους μεταστάσεις όταν ανευρίσκονται, δεν απαντούν στη συστηματική θεραπεία και έχουν πολύ κακή πρόγνωση. Η συντηρητική θεραπεία με ανάλογα σωματοστατίνης ή κετοκοναζόλη δεν έχει κανένα αποτέλεσμα. Μόνο η αμφωτερόπλευρη επινεφριδεκτομή ή η εκτεταμένη R0 εκτομή μπορεί να έχουν κάποιο αποτέλεσμα.
Νευροτενσίνωµα
Η νευροτενσίνη είναι ένα νευροπεπτίδιο που βρίσκεται στον εγκέφαλο και το έντερο. Εμφανίζει ποικίλα συστηματικά αποτελέσματα όταν εκκρίνεται σε υπερβολικά ποσά από έναν όγκο, όπως: ταχυκαρδία, υπόταση, κυάνωση, επίδραση στην εντερική κινητικότητα και την έκκριση εντερικού υγρού καθώς και δράση επί της εξωκρινούς παγκρεατικής εκκρίσεως πρωτεϊνών και διττανθρακικών αλάτων.
Το σύνδρομο υπερπαραγωγής νευροτενσίνης από ένα PNET έχει προταθεί σε διάφορες μελέτες με κλινικά σημεία την υποκαλιαιμία, διάρροια, την απώλεια βάρους, το σακχαρώδη διαβήτη, την κυάνωση, την υπόταση και το flushing. Επειδή οι ασθενείς αυτοί είναι πολύ σπάνιοι, υπάρχει διαφωνία για την ύπαρξη ενός τέτοιου ξεχωριστού συνδρόμου, μια και οι όγκοι αυτοί εκκρίνουν συχνά νευροτενσίνη, παράλληλα με άλλες ορμόνες, όπως VIP ή γαστρίνη. Τα νευροτενσινώματα είναι σπάνιοι όγκοι (έχουν ανακοινωθεί 50 περιπτώσεις), συνήθως κακοήθεις. Η R0 εκτομή μπορεί να είναι θεραπευτική.
PPόµα
Το PPόμα είναι ένας όγκος συνήθως του παγκρέατος που εκκρίνει υπερβολικά ποσά PP (pancreatic polypeptide). Τα κλινικά συμπτώματά του οφείλονται στα τοπικά αποτελέσματα που έχει η μάζα του όγκου και όχι στη δράση του PP. Έχει αναφερθεί σε ασθενείς με PPώματα υδαρής διάρροια με εξάνθημα δέρματος.
Οι όγκοι αυτοί συνήθως δεν συνδέονται με κλινικό σύνδρομο οφειλόμενο σε υπερέκκριση ορμόνης και γι’ αυτό πολλοί τα κατατάσσουν στα NF-pNETs. Τα PPόματα είναι σχεδόν πάντα μεγάλα – εκτός από αυτά που ανευρίσκονται σε ασθενείς με το σύνδρομο ΜΕΝ I – είναι συνήθως μονήρη και εντοπίζονται στην κεφαλή του παγκρέατος. Περίπου το 2/3 είναι κακοήθη, η δε 5ετής και 10ετής επιβίωση είναι περίπου 65% και 49%, αντίστοιχα. Η διάγνωση τίθεται με την ανοσοϊστοχημεία στο ιστολογικό παρασκεύασμα ενός pNET σε συνδυασμό με την τιμή του PP και αυτές των άλλων ορμονών στο αίμα.
Παρατηρούνται υψηλά επίπεδα PP συχνά και σε άλλα λειτουργικά pNETs (22-71%), αλλά και σε μη παγκρεατικά καρκινοειδή. Έτσι υψηλά επίπεδα PP πλάσματος δεν θεμελιώνουν τη διάγνωση ενός PPόμματος ακόμα και επί υπάρξεως παγκρεατικής μάζας. Επιπλέον, υψηλά επίπεδα PP πλάσματος μπορούν να εμφανιστούν και σε άλλες καταστάσεις όπως σε ηλικιωμένους ασθενείς, μετά από εντερεκτομή, με την κατάχρηση οινοπνεύματος, κατά τη διάρκεια ορισμένων λοιμώξεων και άλλων φλεγμονωδών νόσων, σε οξεία διάρροια, χρόνια νεφρική ανεπάρκεια, σε σακχαρώδη διαβήτη κ.λπ. Ίσως μια δοκιμασία με ατροπίνη μπορεί να αποδειχθεί χρήσιμο εργαλείο στη διαφορική διάγνωση τέτοιων καταστάσεων.
Άλλα pNETs
Καρκινοειδές παγκρέατος
Υπάρχουν ακόμα πιο σπάνια pNETς, όπως είναι τα καρκινοειδή του παγκρέατος (όγκοι που παράγουν σεροτονίνη) που είναι συνήθως μεγάλοι (5-6 cm). Μέχρι το 2004, είχαν περιγραφεί 165 περιπτώσεις παγκοσμίως. Μια περίπτωση έχει αναφερθεί να συνδέεται με ΜΕΝ Ι.
Είναι μεγάλα νεοπλάσματα, πολύ συχνά κακοήθη με μεταστάσεις στο 66% περίπου. Καρκινοειδές σύνδρομο ανευρίσκεται σε ποσοστό σχετικά υψηλό για καρκινοειδή (23,3%). Εκφράζουν με ανοσοϊστοχημεία σεροτονίνη στο 93% περίπου, με ανεύρεση 5-υδροξυτρυπταμίνης (5-HT) στα ούρα στο 84% των ασθενών αυτών, ενώ όλοι οι ασθενείς δεν παρουσιάζουν αύξηση της σεροτονίνης στο αίμα. Η μόνη θεραπεία είναι η χειρουργική εξαίρεση. Η μεταμόσχευση ήπατος (ΜΗ) είναι μια λύση στη μεταστατική νόσο. Το πενταετές ποσοστό επιβίωσης είναι εξαιρετικά χαμηλό (28,9%) έναντι του αντίστοιχου της σκωληκοειδούς αποφύσεως (89,7%) και του λεπτού εντέρου (82,1%).
Ακόμη πιο σπάνια pNETs είναι αυτά που εκκρίνουν parathyroid hormone-related peptide (PTH-rP) (parathyrinomas – έχουν ανακοινωθεί 35 περιπτώσεις) και προκαλούν υπερασβεστιαιμία και υπερπαραθυρεοειδισμό. Η πρωτεΐνη αυτή είναι γνωστό ότι προκαλεί υπερασβεστιαιμία σε πολλούς κακοήθεις όγκους που την παράγουν στα πλαίσια του παρανεοπλασματικού συνδρόμου με μηχανισμό παρόμοιο της PTH. Οι όγκοι αυτοί είναι συνήθως μεγάλοι και συνήθως με μεταστάσεις κατά τη διάγνωση και μπορεί να ανταποκριθούν στην εκτομή και τη χημειοθεραπεία.
Πάρα πολύ σπάνια έχουν αναφερθεί pNETs που εκκρίνουν καλτσιτονίνη, εντερογλουκαγόνο, χολοκυστοκίνη, GIP (gastric inhibitory peptide), LH (Luteinizing hormone), GRP (gastrin-releasing peptide) και γρελίνη.

